amplimap documentation¶
A mapping and analysis pipeline for targeted NGS data (version 0.4.20)¶
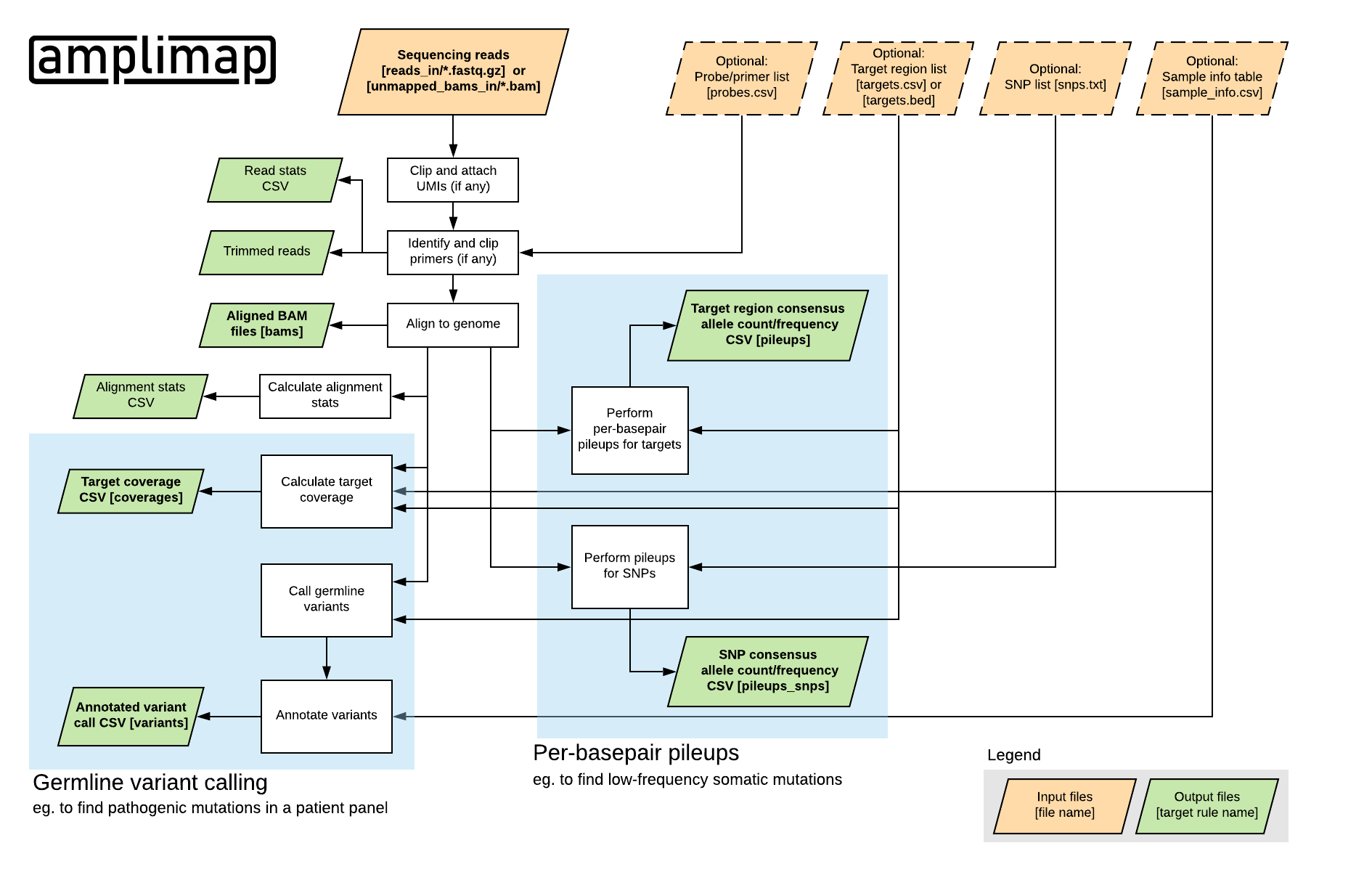
amplimap is a command-line tool to automate the processing and analysis of data from targeted next-generation sequencing (NGS) experiments with PCR-based amplicons or capture-based enrichment systems.
From raw sequencing reads, amplimap generates a variety of output files including read alignments, per-basepair nucleotide counts, target coverage data and annotated variant calls.
In addition to its focus on user-friendliness and reproducibility, amplimap supports advanced features such as the generation of consensus base calls for read families based on molecular identifiers/barcodes (UMIs) and the detection of chimeric reads caused by amplification of off-target loci.
Overview¶
To run amplimap you create a directory containing a small set of input files (see Input and the working directory):
- A subdirectory with FASTQ.GZ or BAM files representing your different samples (tested with Illumina MiSeq, HiSeq and NextSeq)
- Optionally: Files describing the targeted genomic regions, the primers you used or other custom configuration parameters
Then you can run amplimap to generate a variety of different output files, depending on your experiment.
These include, for example:
- A target coverage table, showing you how well-covered each target region was in each sample.
- A table of germline variants in your samples, annotated with gene, impact, population frequencies, deleteriousness scores, etc.
- A per-basepair “pileup” table telling you how often each nucleotide was seen in each sample at each position.
Built on top of Snakemake and Python 3, amplimap is entirely automated and can be run on a single machine as well as on an HPC cluster (e.g. LSF, SGE).
Supported experimental protocols¶
amplimap is compatible with most targeted sequencing protocols that generate paired-end short read data.
For protocols utilising PCR or smMIPs each read should start with a known primer (or targeting arm) sequence, followed by the amplified target DNA. Reads can optionally contain a unique molecular identifier (UMI) sequence in front of the primer, which can be used to group reads into families. Data should be available as demultiplexed FASTQ.GZ files, with each pair of files representing a different sample.
For capture-based protocols data can be provided in FASTQ.GZ or unmapped/mapped BAM format, which may contain UMIs as BAM tags. See Running on capture-based data for details.
Some of the protocols we have analyzed with amplimap include:
- PCR-based targeted resequencing (single/multiplex)
- smMIPs with and without UMIs
- Probe based target enrichment, for example:
Documentation contents¶
Links¶
- Package: https://pypi.org/project/amplimap/
- Code: https://github.com/koelling/amplimap/
- Documentation: https://amplimap.readthedocs.io/
Citation and License¶
Licensed under the Apache License, version 2.0. Copyright 2020 Nils Koelling. When you use amplimap, please cite the amplimap paper in your work:
Nils Koelling, Marie Bernkopf, Eduardo Calpena, Geoffrey J Maher, Kerry A Miller, Hannah K Ralph, Anne Goriely, Andrew O M Wilkie, amplimap: a versatile tool to process and analyze targeted NGS data, Bioinformatics, Volume 35, Issue 24, 15 December 2019, Pages 5349–5350, https://doi.org/10.1093/bioinformatics/btz582